Reaction Details  Report a problem with these data
Report a problem with these data
Report a problem with these dataTarget
Alpha-1D adrenergic receptor
Ligand
BDBM50026917
Substrate
n/a
Meas. Tech.
ChEBML_1691075
Ki
1.3±n/a nM
Citation
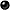 Del Bello, F; Bonifazi, A; Giannella, M; Giorgioni, G; Piergentili, A; Petrelli, R; Cifani, C; Micioni Di Bonaventura, MV; Keck, TM; Mazzolari, A; Vistoli, G; Cilia, A; Poggesi, E; Matucci, R; Quaglia, W The replacement of the 2-methoxy substituent of N-((6,6-diphenyl-1,4-dioxan-2-yl)methyl)-2-(2-methoxyphenoxy)ethan-1-amine improves the selectivity for 5-HT Eur J Med Chem 125:233-244 (2017) [PubMed] Article
Del Bello, F; Bonifazi, A; Giannella, M; Giorgioni, G; Piergentili, A; Petrelli, R; Cifani, C; Micioni Di Bonaventura, MV; Keck, TM; Mazzolari, A; Vistoli, G; Cilia, A; Poggesi, E; Matucci, R; Quaglia, W The replacement of the 2-methoxy substituent of N-((6,6-diphenyl-1,4-dioxan-2-yl)methyl)-2-(2-methoxyphenoxy)ethan-1-amine improves the selectivity for 5-HT Eur J Med Chem 125:233-244 (2017) [PubMed] ArticleMore Info.:
Target
Name:
Alpha-1D adrenergic receptor
Synonyms:
ADA1D_HUMAN | ADRA1A | ADRA1D | Adrenergic receptor | Adrenergic receptor alpha | Alpha 1D-adrenoceptor | Alpha 1D-adrenoreceptor | Alpha adrenergic receptor (1a and 1d) | Alpha-1D adrenoceptor | Alpha-adrenergic receptor 1a | adrenergic Alpha1D
Type:
Enzyme Catalytic Domain
Mol. Mass.:
60485.82
Organism:
Homo sapiens (Human)
Description:
adrenergic Alpha1D ADRA1D HUMAN::P25100
Residue:
572
Sequence:
MTFRDLLSVSFEGPRPDSSAGGSSAGGGGGSAGGAAPSEGPAVGGVPGGAGGGGGVVGAGSGEDNRSSAGEPGSAGAGGDVNGTAAVGGLVVSAQGVGVGVFLAAFILMAVAGNLLVILSVACNRHLQTVTNYFIVNLAVADLLLSATVLPFSATMEVLGFWAFGRAFCDVWAAVDVLCCTASILSLCTISVDRYVGVRHSLKYPAIMTERKAAAILALLWVVALVVSVGPLLGWKEPVPPDERFCGITEEAGYAVFSSVCSFYLPMAVIVVMYCRVYVVARSTTRSLEAGVKRERGKASEVVLRIHCRGAATGADGAHGMRSAKGHTFRSSLSVRLLKFSREKKAAKTLAIVVGVFVLCWFPFFFVLPLGSLFPQLKPSEGVFKVIFWLGYFNSCVNPLIYPCSSREFKRAFLRLLRCQCRRRRRRRPLWRVYGHHWRASTSGLRQDCAPSSGDAPPGAPLALTALPDPDPEPPGTPEMQAPVASRRKPPSAFREWRLLGPFRRPTTQLRAKVSSLSHKIRAGGAQRAEAACAQRSEVEAVSLGVPHEVAEGATCQAYELADYSNLRETDI
Inhibitor
Name:
BDBM50026917
Synonyms:
8-(2-(4-(2-methoxyphenyl)piperazin-1-yl)ethyl)-8-azaspiro[4.5]decane-7,9-dione | 8-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-8-azaspiro[4.5]decane-7,9-dione | 8-{2-[4-(2-Methoxy-phenyl)-piperazin-1-yl]-ethyl}-8-aza-spiro[4.5]decane-7,9-dione | 8-{2-[4-(2-Methoxy-phenyl)-piperazin-1-yl]-ethyl}-8-aza-spiro[4.5]decane-7,9-dione(BMY-7378) | BMY 7378 | BMY-7378 | CHEMBL1256934 | CHEMBL13647 | CHEMBL543741
Type:
Small organic molecule
Emp. Form.:
C22H31N3O3
Mol. Mass.:
385.4998
SMILES:
COc1ccccc1N1CCN(CCN2C(=O)CC3(CCCC3)CC2=O)CC1
Structure: